CH391L/NucleotideFrequency
From Marcotte Lab
Source Code
How it works
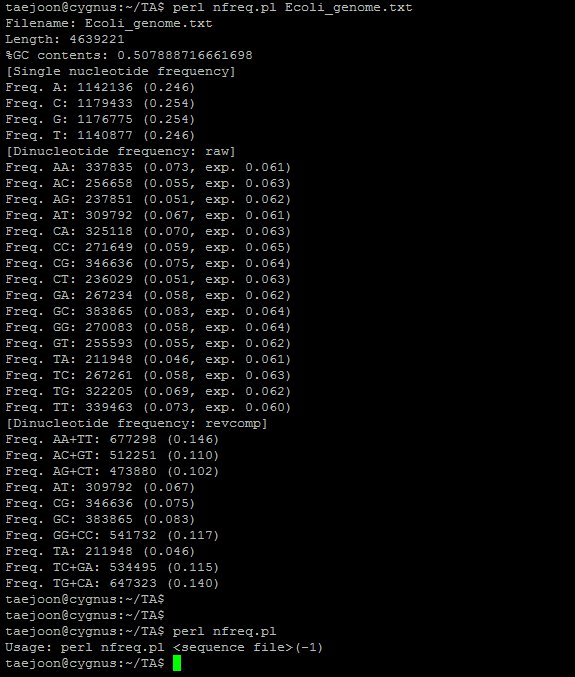
Comments
- For dinucleotide frequency, I considered two cases separately: raw and revcomp. 'raw' case is straightforward, just count all dinucleotides available on given sequences with overlap. 'revcomp' case is little bit tricky, because it takes the other strand sequence into account (remember the given sequence is one strand of double helix DNA molecule, so there's complementary strand available). In single nucleotide frequency, we normally compare 'A+T' contents and 'G+C' contents, because amount of 'G' in a given strand is same as the amount of 'C' in complementary strand (same as A and T). You can expand this idea to dinucleotide frequency. However, some dinucleotides, such as 'AT' or 'CG', have exactly same reverse complementary dinucleotide (palindromic sequence), so I handled them separately.
- The following expressions are identical. I personally prefer to use the second one, because it is concise, more flexible than third one (i.e. add by 10 instead of 1). Also, some programming languages, i.e. python, do not support third.
$a = $a + 1; $a += 1; $a++;
- Although it is not necessary, it is a good programming habit to initialize your variable. Use '%freq = ();' instead of '%freq;'.
- Ignore 'my' on my code if you did not use 'strict' and '-w'. We did not cover this in the class, and PERL works fine without them. I used them for a long time, so I am little bit nervous without using these. If you are curious what they are, and why I have used them, check here.